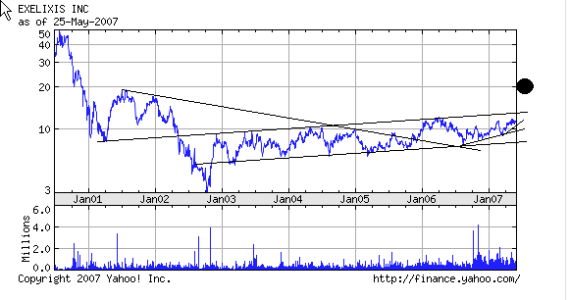
") www.exelixis.com
www.exelixis.com.
At Exelixis, our commitment to improving the treatment of patients with
cancer has driven us to pursue an uncommon path and a different approach to developing drugs. We have built critical mass throughout all areas of our research and development infrastructure, making it possible to achieve unparalleled productivity while retaining an unwavering commitment to quality. Our biology-based and data-driven approach is designed to enable the development of first-in-class or best-in class compounds that will potentially improve the care and outcomes of cancer patients. Our unique strategy is designed to ensure a steady stream of highly qualified compounds that address the changing needs of cancer patients.
Recognizing that today’s cancer patients continue to have significant unmet medical needs, we act with a sense of urgency that mandates that we think large and move fast, pursuing a better way to better medicine.
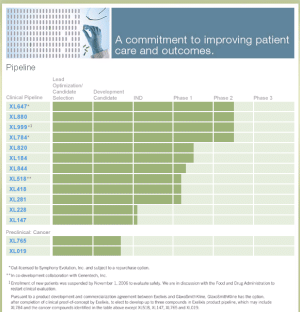
Spectrum Selective Kinase Inhibitors
Our first generation clinical development candidates are each designed to simultaneously target multiple members of a family of proteins known as receptor tyrosine kinases (RTKs). These proteins are involved in key cancer-related processes, including cell growth and proliferation, survival, metastasis, tumor angiogenesis and drug resistance. RTK inhibition has been validated as a therapeutic approach to treating cancer by the approval of numerous drugs designed to target specific RTKs.
The compounds that are developing are designed to target multiple RTKs simultaneously have been exhaustively optimized for tolerability, potency, specificity, half-life and dosing schedule against each of their multiple targets, which may provide improved efficacy and enhanced safety profiles compared with combinations of single-target drugs that have not been optimized for use together. This approach may provide a way to inhibit numerous cancer processes – such as angiogenesis, cell growth or drug resistance – in a highly concerted manner with a single drug. In turn, this may achieve a clinically meaningful balance between the broad effects of chemotherapy and the favorable safety and tolerability profiles of targeted agents.
Inhibiting Critical Downstream Pathways
Recent breakthroughs in molecular biology have identified a number of critical downstream signaling pathways that are essential for translating the activity of cancer-related RTKs into malignant processes. Our second generation compounds are focused on inhibiting critical downstream pathways such as PTEN/PI3K, RAS/RAF/MEK/ERK and JAK/STAT. These pathways regulate growth and survival and are mutationally activated in many cancers. Individual kinases within these pathways are points of convergence for multiple signaling pathways. Thus, inhibiting one of these downstream kinases could effectively shut down signaling through a number of different pathways.
We are leveraging our expertise in comparative genomics and molecular biology to explore the complex interactions among an array of upstream and downstream signaling processes. We believe that a comprehensive understanding of the roles that these pathways play in cancer will enhance our ability to select targets with the potential to enable entirely new classes of cancer therapies. Already we have made significant progress in developing a portfolio of compounds that inhibit critical components of downstream signaling pathways with high specificity and activity
......................................................................................................................................................................................
XL647
XL647 is a potent inhibitor of RTKs that are implicated in driving tumor proliferation and angiogenesis (tumor blood vessel formation). XL647 inhibits the EGFR, HER2 and VEGFR RTKs simultaneously in preclinical studies. Preliminary data from a Phase I trial of XL647 were presented in November 2005, and at the American Society of Clinical Oncology (ASCO) annual meeting in June 2006. Updated data were presented in November 2006 at the 18th EORTC-NCI-AACR Symposium on Molecular Targets and Cancer Therapeutics.
As of August 1, 2005, 41 patients had been enrolled in the trial and were evaluable for safety and tumor response assessments. Investigators report that one patient with a primary diagnosis of NSCLC had achieved a partial response (unconfirmed) that occurred prior to cycle 16 (approximately 7.5 months of treatment) and then developed progressive disease prior to cycle 20. The patient received a total of 19 cycles (9.5 months). Fourteen additional patients have had stable disease for at least three months (NSCLC [3], chordoma [2], adenoid cystic [2], head and neck [2] and one each with adrenocorticoid, mesothelioma, colorectal, ovarian and leiomyosarcoma).
As reported previously by investigators, the most common treatment-related adverse events seen in the XL647 clinical trial as of August 1, 2005 were grade 1 or 2 rash, diarrhea, nausea, or fatigue. Hematologic toxicities (9.5% of all toxicities), anemia (7.1%) and grade 2 thrombocytopenia (2.4%) were also reported. Two events considered possibly related to study treatment have been reported: a grade 4 pulmonary embolism and a grade 3 elevation in INR in a patient taking concomitant warfarin. Two patients enrolled experienced grade 3 diarrhea that was considered probably related to study treatment. Three dose-limiting toxicities occurred: a grade 3 QTc prolongation (probably related) and two incidences of grade 3 diarrhea.
A Phase II trial of XL647 in patients with advanced (stage IIIB or IV) non-small cell lung cancer (NSCLC) who have not previously been treated with chemotherapy was initiated in August 2006. Participants must meet at least two of the following criteria: asian, female, non-smoker or adenocarcinoma. The multi-center, open-label Phase II study will be conducted in up to 15 clinical sites and will follow a two-stage enrollment strategy. The primary objectives of the Phase II study are to determine the response rate of subjects with NSCLC treated with XL647 and to evaluate the safety and tolerability of XL647. Secondary objectives include assessment of progression-free survival, duration of response, and overall survival, and characterization of pharmacokinetic and pharmacodynamic parameters of XL647. Additionally, we are considering combination trials of XL647 with other anticancer treatments to test the ability of the combination therapy to prolong progression free survival.
Poster: XL647 AACR-NCI-EORTC (November 2006)
Poster: XL647 ASCO (June 2006)
Poster: XL647 AACR-NCI-EORTC (November 2005)
< top
XL880
XL880 inhibits Met and VEGFR2, which play synergistic roles in promoting tumor growth and angiogenesis. Activation or overexpression of Met is a prevalent feature of a wide spectrum of human tumors and is a negative prognostic indicator in patients with multiple myeloma, glioma and certain solid tumors. XL880 is the first small molecule Met inhibitor to enter the clinic. Interim data from an ongoing Phase I study of XL880 were presented in November 2005, and at the American Society of Clinical Oncology (ASCO) annual meeting in June 2006. Most recently, data from this study were reported at the 18th EORTC-NCI-AACR Symposium on Molecular Targets and Cancer Therapeutics in November 2006.
As of October 6, 2006, 40 patients had been enrolled in the Phase I trial and were evaluable for safety; and 29 of which were also evaluable for pharmacokinetic analyses. As reported by the investigators, four patients have had partial responses (papillary renal cell carcinoma [3] and Hurthle cell carcinoma [1]); four patients have had minimal responses (carcinoid [1], colorectal [1], melanoma [1] and medullary thyroid [1]); and seven patients have had stable disease for 3+ to 7 months (colorectal [3], renal cell [1], billiary [1], urethral [1] and thyroid [1]).
With respect to biomarkers for XL880 activity, analyses of melanoma, breast and medullary thyroid tumor samples indicate that XL880 decreases the phosphorylation status of Met, RON, ERK, and AKT, decreases Ki67, a marker for tumor cell proliferation, and increases apoptosis of tumor and endothelial cells. This effect was not observed in samples of normal tissue obtained at the same time points.
Two serious adverse events in the maximum administered dose (MAD) cohort of 4.5 mg/kg that were reported as possibly and probably related to study drug included a grade 3 tumor hemorrhage and a grade 3 hand/foot syndrome. One serious event of confusion, altered speech or "expressive language disorder" was reported for one patient in the MTD cohort at the 3.6 mg/kg dose. The event was grade 2, fully reversible, and the patient continues on study with minimal response at 2.4 mg/kg and no recurrence of symptoms. Grade 2 or greater adverse events considered possibly or probably related to XL880 treatment in at least two patients included fatigue (grade 2, two patients) and hypertension (grade 2, two patients). Hypertension is a dose-related side effect.
A Phase II clinical development program for XL880 was initiated in June 2006 in patients with papillary renal cell carcinoma. The multi-center open-label Phase II study will be conducted in up to 15 clinical sites and is designed to enroll up to 34 patients with hereditary or sporadic papillary renal cell carcinoma. Primary objectives of the study are to determine best-confirmed response rate and to evaluate safety and tolerability of XL880 administered orally for five consecutive days every two weeks. Secondary objectives are to assess progression-free survival, overall survival, duration of response and to continue characterizing the pharmacokinetic and pharmacodynamics profiles of XL880.
The multi-center open-label Phase II study in gastric cancer was initiated in December 2006 and will be conducted at multiple clinical sites and enroll patients with metastatic, poorly differentiated diffuse gastric cancer, a tumor type that is associated with amplification of the MET gene. The primary objectives of the study are to determine best- confirmed response rate and to evaluate safety and tolerability of XL880 administered orally for five consecutive days every two weeks. Secondary objectives are to assess progression-free survival, overall survival, duration of response, and to continue characterizing the pharmacokinetic and pharmacodynamic profiles of XL880. Subsequent Phase II trials are planned in head and neck cancer.
Poster: XL880 AACR-NCI-EORTC (November 2006)
Poster: XL880 ASCO (June 2006)
Poster: XL880 AACR-NCI-EORTC (November 2005)
< top
XL999
XL999is a potent inhibitor of key receptor tyrosine kinases (RTKs) implicated in the development and maintenance of tumor vasculature and in the proliferation of some tumor cells. It inhibits the FGFR, VEGFR and PDGFR RTKs and also is a potent inhibitor of FLT3, an important driver of leukemia cell proliferation in some patients with acute myelogenous leukemia (AML).
In the third quarter of 2006 we received new data that impacted the Phase II trials for XL999. Although there were encouraging signs that XL999 has the potential to provide benefit to patients with lung cancer and acute myelogenous leukemia our internal safety monitoring board became concerned with the frequency of cardiovascular events experienced in October by patients in the program, and by November 1, we suspended enrollment in all XL999 trials pending collection and analysis of further data. We determined that patients already enrolled in the Phase I and II trials could continue to receive XL999 and the U.S. Food and Drug Administration (FDA) later agreed with this determination. After extensive review of the data, we submitted a proposed action plan to the FDA in January 2007, and we are working with the FDA to restart clinical evaluation. We believe that XL999 has the potential to provide a risk-to-benefit profile that may make this compound an attractive treatment option for patients with a variety of cancers.
Poster: XL999 AACR-NCI-EORTC (November 2006)
Poster: XL999 AACR-NCI-EORTC (November 2005)
< top
XL784
XL784 was the first small molecule compound developed using our proprietary drug discovery engine. The compound is a potent inhibitor of the ADAM-10 metalloprotease enzyme, a target of significant interest because of its important role in blood vessel formation and cell proliferation. XL784 was specifically optimized to be matrix metalloprotease-1 (MMP-1) sparing, thus potentially significantly enhancing its safety profile and enabling higher dosing compared with other previously studied metalloprotease inhibitors. Results of a single dose Phase I clinical trial of XL784 administered orally to 70 healthy volunteers demonstrated that XL784 has attractive safety and pharmacokinetic profiles.
A repeat-dose Phase I clinical trial of a capsule formulation of XL784 was completed in healthy volunteers in 2005 and a Phase II double-blind, placebo-controlled trial in patients with proteinuria associated with diabetic nephropathy is ongoing.
< top
XL820
XL820 has demonstrated potent inhibitory activity in preclinical models against KIT, VEGFR and PDGFR, clinically validated targets implicated in a variety of human cancers. In cellular models, XL820 is a potent inhibitor of mutationally activated forms of KIT found in human cancers. In tumor models of breast carcinoma, glioma and leukemia the compound exhibited dose-dependent growth inhibition and has been shown to cause tumor regression. A Phase I clinical trial of XL820 was initiated in July of 2005 in patients with solid tumors for whom there are no available therapies known to prolong survival. Preliminary data from this study were reported at the 18th EORTC-NCI-AACR Symposium on Molecular Targets and Cancer Therapeutics in November 2006.
As of September 22, 2006, 23 patients had been enrolled in the trial. Patients received XL820 orally on an intermittent schedule with 5 daily doses given every 2 weeks. Twenty-two were evaluable for safety assessments and 19 were evaluable for tumor response. As reported by the investigators, four patients have had stable disease for 3.5+ to 11+ months (one each with gastrointestinal stromal tumor [GIST], renal cell carcinoma, testicular cancer and thyroid cancer). One instance of dose-limiting toxicity has been reported (grade 3 elevation of aspartate aminotransferase); no grade 4 adverse events considered related to XL820 have been reported to date
Poster: XL820 AACR-NCI-EORTC (November 2006)
< top
XL184
XL184 inhibits VEGFR and MET, key drivers for tumor formation and growth. The compelling preclinical efficacy of XL880, our first VEGFR2/Met inhibitor, increased our interest in inhibitors of these RTKs and resulted in the discovery and development of XL184, a highly potent VEGFR2 inhibitor with nanomolar potency against Met. XL184 has demonstrated potent growth inhibition and tumor regression in a variety of tumor models. A Phase I clinical trial in patients with solid tumors for whom there are no available therapies was initiated in September 2005. Preliminary data from this study were reported at the 18th EORTC-NCI-AACR Symposium on Molecular Targets and Cancer Therapeutics in November 2006.
As of October 6, 2006, 21 patients had been enrolled in the trial, of which 18 were evaluable for safety and pharmacokinetic analyses. To date, there have been six XL184-related adverse events reported. None of these events was dose limiting and further dose escalation is currently ongoing. The pharmacokinetic profile of XL184 showed a long half-life of 59.1 - 98.3 hours. This was independent of dose and duration of treatment. As reported by the investigators, there were three patients with some improvement in disease measures. One patient with carcinoid carcinoma had a reported decrease in tumor size (20%), one patient with cutaneous T cell lymphoma had improvement in skin lesions, and one patient with medullary thyroid carcinoma had a decrease in serum tumor markers. Three additional patients have achieved prolonged stable disease of at least three months (carcinoid carcinoma [2] and metastatic parotid tumor [1]).
Poster: XL184 AACR-NCI-EORTC (November 2006)
< top
XL844
XL844 potently inhibits CHK1 and CHK2, kinases that induce cell cycle arrest in response to a variety of DNA damaging agents, allowing repair of damaged DNA and promoting resistance to many standard chemotherapies. In preclinical studies, XL844 significantly enhances the ability of multiple chemotherapeutic agents to kill tumor cells without increasing systemic toxicity. A Phase I clinical trial of XL844 in patients with chronic lymphocytic leukemia was initiated in September 2005.
< top
XL518
XL518 is a novel small molecule drug designed to inhibit the activity of MEK, a key component of the RAS/RAF/MEK/ERK signaling pathway. This pathway is frequently activated in human tumors, and is required for transmission of growth-promoting signals from numerous receptor tyrosine kinases. Preclinical studies have demonstrated that XL518 is a potent and specific inhibitor of MEK, with highly optimized pharmacokinetic and pharmacodynamic properties. XL518 has excellent oral bioavailability in multiple species, and induces substantial and durable inhibition of ERK phosphorylation in xenograft tumor models. Administration of XL518 causes tumor regression in multiple xenograft models with mutationally-activated B-RAF or RAS. A Phase I clinical trial initiated in April 2007.
< top
XL418
XL418 is a small molecule that inhibits the activity of protein kinase B (PKB or AKT) and S6 Kinase (S6K), which act downstream of phosphoinosotide-3 kinase (PI3K) and is the first of three internally generated compounds that target the PI3K/PTEN pathway. Activation of these kinases is a frequent event in human tumors, promoting cell growth, survival, and resistance to chemotherapy and radiotherapy. Inactivation of the pathway through inhibition of AKT is expected to induce apoptosis (programmed cell death) in tumor cells. AKT inhibitors may also sensitize tumor cells to a wide range of chemotherapy. In preclinical studies, XL418 slowed tumor growth in multiple cancer models, including breast and lung adenocarcinomas. XL418 also has been shown to enhance apoptosis in combination with XL647, an inhibitor of multiple receptor tyrosine kinases including EGFR, HER2, and VEGFR, in preclinical tumor models. A Phase I clinical trial initiated in April 2007.
< top
XL281
XL281 is a novel small molecule drug designed to specifically inhibit RAF kinases, which lie immediately downstream of RAS and are key components of the RAS/RAF/MEK/ERK kinase signaling pathway. Genetic lesions that activate this pathway are common in human tumors, with activating mutations in K-RAS occurring in 30 percent of tumors and activating mutations in B-RAF occurring in approximately 60 percent of melanomas. The RAS/RAF/MEK/ERK pathway also plays a key role in the transmission of growth-promoting signals downstream of receptor tyrosine kinases. This suggests that deregulation of this pathway plays a pivotal role in the progression of many human tumors, and that inhibition of the pathway may provide clinical benefit in the treatment of cancer. In preclinical studies, XL281 showed potent inhibition of B-RAF, mutationally activated B-RAF and C-RAF, and did not interact with kinases outside of the RAF family. XL281 displays high oral bioavailability and strongly inhibits RAS/RAF/MEK/ERK signaling in human tumor models. This translates into substantial inhibition of tumor growth in preclinical xenograft models of human tumors that overexpress receptor tyrosine kinases or harbor activating mutations in RAS or RAF. A Phase I clinical trial initiated in April 2007.
< top
XL228
XL228 is a potent inhibitor of several tyrosine kinases implicated in the growth, proliferation and metastasis of cancer cells. The compound inhibits the activity of the insulin-like growth factor type-1 receptor (IGF1R), SRC and BCR-ABL. Significantly, in preclinical studies XL228 had potent activity against the T315I mutant form of Abl, which is associated with resistance to currently approved therapies. In addition, administration of XL228 resulted in significant tumor growth inhibition and regression in xenograft tumor models. An IND for XL228 was filed in August 2006, and a Phase I clinical trial of XL228 is expected to initiate in 2007.
< top
XL147
XL147 is an orally available small molecule that selectively inhibits the activity of phosphoinositide-3 kinase (PI3K). XL147 is the second of three internally generated compounds that target the PI3K/PTEN pathway. Activation of PI3K is a frequent event in human tumors, promoting tumor cell growth, survival, and resistance to chemotherapy and radiotherapy. Inactivation of PI3K has been shown to inhibit growth and induce apoptosis (programmed cell death) in tumor cells. In preclinical studies, XL147 slowed tumor growth or caused tumor shrinkage in multiple preclinical cancer models, including breast, lung, ovarian, and prostate cancers, and gliomas. XL147 has also been shown to enhance the anti-tumor effects of several chemotherapeutic agents and an inhibitor of epidermal growth factor receptor (EGFR) in preclinical cancer models. An IND for XL147 was filed in March 2007, and a Phase I clinical trial is expected to initiate in 2007.
< top
......................................................................................................................................................................................
Preclinical: Cancer
The current preclinical oncology pipeline includes inhibitors of: PI3K/mTOR (XL765) and JAK2 (XL019). These compounds have been designated as a potential drug candidate and will potentially support the filing of an IND in 2007.
PI3K/mTOR (XL765): an orally available small molecule that inhibits the activity of phosphoinositide-3 kinase (PI3K) and mammalian target of rapamycin (mTOR). XL765 is the second of three internally generated compounds that target the PI3K/PTEN pathway. Activation of PI3K is a frequent event in human tumors, promoting tumor cell growth, survival, and resistance to chemotherapy and radiotherapy. mTOR is also frequently activated in human tumors and plays a central role in tumor cell growth. mTOR can be activated via upregulation of PI3K, or via PI3K-independent mechanisms. Inactivation of PI3K has been shown to inhibit growth and induce apoptosis (programmed cell death) in tumor cells, whereas inactivation of mTOR has been shown to inhibit the growth of tumor cells. In preclinical studies, XL765 slowed tumor growth or caused tumor shrinkage in multiple preclinical cancer models, including breast, lung, ovarian, and prostate cancers, and gliomas. XL765 has also been shown to enhance the anti-tumor effects of several chemotherapeutic agents in preclinical cancer models. An IND for XL765 is expected to be filed in 2007.
JAK2 (XL019): Receptor tyrosine kinases and cytokine receptors activate downstream signaling through the JAK pathway, leading to phosphorylation of STAT proteins which subsequently bind DNA, initiating the transcription of target genes regulating cell growth and survival. Activating mutations of JAK2 occur frequently in a variety of myeloproliferative disorders, and JAK2 signaling is upregulated in several types of lymphoma, non-small cell lung cancer, hepatocellular carcinoma, multiple myeloma and prostate cancer. An IND for XL019 is expected to be filed in 2007.
We anticipate multiple new oncology compounds to enter preclinical development in 2007.